
Die ZSVA-Verantwortlichen sind auch für die Technologiefrüherkennung in ihrem Fachbereich zuständig. Zu dieser Pflicht gehört, dass sie die an den Normen und den verschiedenen Referenzdokumenten vorgenommenen Änderungen kennen. Dieser Artikel soll Ihnen helfen, einen Überblick über die Neuerungen zu gewinnen, die sich auf Ihre Arbeit auswirken könnten.
MepV
Im Mai 2021 wurde eine neue Fassung der MepV veröffentlicht, um diese mit der neuen europäischen Medizinprodukte-Verordnung (MDR) in Übereinstimmung zu bringen.
Nachfolgend werden die wichtigsten Änderungen erläutert:
- Artikel 4 enthält eine Definition der Begriffe «Instandhaltung» und «Aufbereitung».
- Instandhaltung: Massnahmen wie Wartung, Softwareupdates, Inspektion, Reparatur, Vorbereitung zur Erstverwendung sowie Aufbereitung zur Wiederverwendung, zur Erhaltung oder Wiederherstellung des funktionsfähigen Zustandes eines Produkts
- Aufbereitung: Verfahren, dem ein gebrauchtes Produkt unterzogen wird, damit es sicher wiederverwendet werden kann; zu diesem Verfahren gehören Reinigung, Desinfektion, Sterilisation und ähnliche Verfahren, insbesondere das Verpacken, der Transport und die Lagerung, sowie Prüfung und Wiederherstellung der technischen und funktionellen Sicherheit des gebrauchten Produkts.
Die Aufbereitung ist folglich Teil der Instandhaltung. Entsprechend gilt die Instandhaltungspflicht nach Artikel 49 HMG.
- Artikel 20 der alten Fassung ist zu Artikel 71 mit folgendem Wortlaut geworden:
Artikel 71 Instandhaltung
1 Wer Produkte als Fachperson anwendet, sorgt für die vorschriftsgemässe Durchführung der Instandhaltung und der damit verbundenen Prüfungen.
2 Die Instandhaltung hat nach den Grundsätzen eines Qualitätsmanagementsystems zu erfolgen, ist zweckmässig zu organisieren und zu dokumentieren und richtet sich insbesondere:
A. nach den Anweisungen des Herstellers;
B. nach dem Risiko, das dem Produkt und seiner Verwendung eigen ist.
3 Für Produkte mit Messfunktion können Prüfverfahren gemäss der Messmittelverordnung vom 15. Februar 200681 vorgesehen werden.
4 Die Swissmedic kann Vorgaben zu Instandhaltungsmassnahmen machen und veröffentlichen. Diese Vorgaben gelten als Stand von Wissenschaft und Technik.
Zu diesen Vorgaben zählen die von Swissmedic veröffentlichten Leitlinien und insbesondere die Schweizerische Gute Praxis zur Aufbereitung von Medizinprodukten.
- Artikel 19 der alten Fassung ist zu Artikel 72 mit folgendem Wortlaut geworden:
Artikel 72 Aufbereitung
1 Wer als Fachperson ein Produkt verwendet, das zur mehrmaligen Anwendung bestimmt ist, sorgt vor jeder Anwendung für die Prüfung der Funktionsfähigkeit und die vorschriftsgemässe Aufbereitung nach dem Stand von Wissenschaft und Technik unter Berücksichtigung der Anweisungen des Herstellers sowie der Anforderungen an die Hygiene.
2 Für die Aufbereitung sind Verfahren zu verwenden, die geeignet und nach dem Stand von Wissenschaft und Technik validiert sind und deren nachgewiesene Wirksamkeit nachvollziehbar und reproduzierbar im Rahmen eines Qualitätsmanagementsystems gewährleistet ist.
3 Wer Produkte für Dritte aufbereitet, muss:
A. zum aufbereiteten Produkt erklären, dass das Produkt:
- nach den Anweisungen des Herstellers aufbereitet worden ist, oder
- nach einem eigenen Aufbereitungsverfahren aufbereitet worden ist, das gleich sicher und gleich wirksam ist wie das vom Hersteller vorgegebene Verfahren, und diese Gleichwertigkeit mit einer Risikoanalyse und einem Validierungsverfahren nachgewiesen wurde;
B. über ein geeignetes, nach national oder international anerkannten Normen zertifiziertes Qualitätsmanagementsystem verfügen;
C. den Nachweis erbringen, dass die Aufbereitung in zweckmässigen Räumlichkeiten nach den anerkannten Regeln von Wissenschaft und Technik erfolgt und dabei die Anforderungen an die Hygiene eingehalten werden;
D. dokumentieren, dass das Produkt gemäss Buchstabe a aufbereitet worden ist.
4 Die Erklärung nach Absatz 3 Buchstabe a muss die Identifikation des Produkts sowie Name und Adresse des aufbereitenden Betriebs enthalten.
Die Aufbereitung hat also gemäss den Anweisungen des Herstellers und nach dem Stand von Wissenschaft und Technik zu erfolgen.
Die ZSVA muss ihre Tätigkeit gemäss geeigneten und validierten Verfahren durchführen.
- Laut Artikel 73 ist die Aufbereitung von gebrauchten Einmalprodukten und deren Weiterverwendung verboten.
- In Artikel 104 ist das Anbringen des UDI (eindeutiger Produktidentifikator) wie folgt geregelt:
A. für implantierbare Produkte und Produkte der Klasse III: ab dem 26. Mai 2021;
B. für Produkte der Klassen IIa und IIb: ab dem 26. Mai 2023;
C. für Produkte der Klasse I: ab dem 26. Mai 2025;
Jede Einrichtung muss sich überlegen, wie der UDI angebracht werden soll und welche Auswirkungen sich auf die ZSVA ergeben.
Normen
- SN EN 17141: Reinräume und zugehörige Reinraumbereiche – Biokontaminationskontrolle (01.2021)
- Sie ersetzt die Norm SN EN ISO 14698-2.
- Tabelle B.1 in Kapitel B 3. enthält die empfohlenen Grenzen für die Überwachung der mikrobiologischen Kontamination in auf Sauberkeit kontrollierten Bereichen während der Herstellung von Medizinprodukten.
- SN EN ISO 17664-1 und 17664-2: Aufbereitung von Produkten für die Gesundheitsfürsorge – Vom Medizinprodukt-Hersteller bereitzustellende Informationen für die Aufbereitung von Medizinprodukten (10.2021)
- Teil 1: Kritische und semi-kritische Medizinprodukte
- Teil 2: Nicht kritische Medizinprodukte
Die Norm bezieht sich auf Medizinprodukte, die zur Wiederverwendung vorgesehen sind und eine Aufbereitung erfordern, durch die sie von ihrem Zustand «nach dem klinischen Gebrauch» in den Zustand gelangen, «erneut gebrauchsfertig» zu sein. Ferner gilt sie für Medizinprodukte für den Einmalgebrauch, die vor der Anwendung aufbereitet werden müssen und in einem sauberen und/oder desinfizierten Zustand verwendet werden sollen. Beispiel: Knochenschrauben.
Die Norm wurde in zwei Teile aufgetrennt.
- SN EN 285: Sterilisation – Dampf-Sterilisatoren – Gross-Sterilisatoren (10.2021)
- Bei der Definition der Begriffe «nicht kondensierbare Gase» und «gesättigter Wasserdampf» wurde die folgende Anmerkung hinzugefügt: Die Anforderungen an die Dampfdurchdringung und die Definition des Begriffs «gesättigter Dampf» sind aktuell Diskussionspunkte sowohl in CEN/TC 102/WG 5 als auch in ISO/TC 198/WG 3.
- 6.4.4.2 Messketten für Mantel und Dampfversorgung
Druck-Messketten müssen:
- in Kilopascal oder Bar anzeigen;
- über den gesamten Skalenbereich von 0 kPa bis 400 kPa eine Fehlergrenze von ±1,6 % oder besser haben;
- eine in Schritten von maximal 20 kPa eingeteilte Skala aufweisen;
- eine Vorrichtung zur Vor-Ort-Justage aufweisen, ohne das Gerät demontieren zu müssen.
8.1 Dampfdurchdringung
ANMERKUNG 1: Jedes Sterilisationsverfahren mit Dampf ist ein einmaliges Ereignis. Wenn auch eine periodisch durchgeführte Prüfung auf Dampfdurchdringung eine sehr nutzbringende Kontrolle des Geräts darstellt, kann dafür gesorgt werden, dass bei jedem Zyklus eine angemessene Dampfdurchdringung angezeigt wird.
Diese Anmerkung würde in die Richtung zielen, bei jedem Sterilisationszyklus eine Dampfdurchdringungsprüfung durchzuführen.
ANMERKUNG 2: Im Gesundheitswesen hat die Verwendung von Instrumenten mit langen Lumen zugenommen. Bei einigen dieser Instrumente kann die Wirksamkeit der Entlüftung, die bei Prüfungen mit Textilbeladungen festgestellt wurde, nicht ausreichend sein.
Folglich sind die Dampfdurchdringungstests je nach den sterilisierten Chargen auszuwählen.
- SN EN ISO 11138-8: Sterilisation von Produkten für die Gesundheitsfürsorge – Biologische Indikatoren – Teil 8: Methode zur Validierung einer reduzierten Inkubationszeit eines biologischen Indikators (08.2021)
- Eine Inkubationszeit von weniger als sieben Tagen gilt als reduzierte Inkubationszeit (Reduced Incubation Time, RIT).
- Die Norm betrifft die Überwachung von Sterilisationsverfahren mit feuchter Hitze oder mit Ethylenoxid, nicht aber mit verdampftem Wasserstoffperoxid.
Wenn Sie biologische Indikatoren für die Validierung von Verfahren für die Wasserdampfsterilisation verwenden, ist diese Norm auf Sie anwendbar. Sie müssen sicherstellen, dass die von Ihnen gekauften Indikatoren diesem neuen Teil der Norm SN EN ISO entsprechen.
- SN EN ISO 15883-5: Reinigungs-Desinfektionsgeräte – Teil 5: Leistungsanforderungen und Kriterien für Prüfverfahren zum Nachweis der Reinigungswirksamkeit (10.2021)
- Sie ersetzt die Fassung von 2005.
- Bei den Begriffen sind zwei Definitionen wichtig:
- Rein: frei von Anschmutzungen und unterhalb festgeschriebener Level von Analyten
- Analyt: chemische Substanz, die Gegenstand einer chemischen Analyse ist
- 4.1.5: Prüfung der Reinigungswirksamkeit: Die Prüfung auf Leistungsqualifizierung ist mit den ungünstigsten Beladungen, die durch die klinische Anwendung angeschmutzt sind, oder mit Ersatzprodukten, sofern dies gerechtfertigt ist, durchzuführen.
Es wird empfohlen, routinemässig angeschmutztes Material zu benutzen. Vorgeschrieben ist dies allerdings nicht. Eine kritische Charge kann auch aus Ersatzprodukten mit geeigneter Prüfanschmutzung bestehen.
- 4.2: Prüfanschmutzungen: Die Begründung für die Wahl der Prüfanschmutzung(en) und des (der) Anschmutzungsverfahren(s) muss begründet und dokumentiert werden. Die Rezepturen der Prüfanschmutzung dürfen auf Grundlage des Studiums der Literatur und des Nachweises ihrer Relevanz gewählt oder entwickelt werden.
- Tabelle A.1 im A enthält Beispiele je nach chirurgischem Verfahren.
Die Wahl der Prüfanschmutzungen (Leistungsqualifizierung und Routineprüfungen) muss folglich wissenschaftlich begründet und dokumentiert werden.
- 4.2.4: Das Verfahren für die Extraktion der Prüfanschmutzung, die Wirksamkeit der Rückgewinnung sowie der Nachweis von Analyten müssen validiert und festgelegt werden. Die Validierung der Rückgewinnung muss die Fähigkeit zur Reduktion des Analyten unter die Eingriffsgrenze nachweisen. Eine angemessene prozentuale Rückgewinnung beträgt mehr als 70 %, sofern nicht anderweitig begründet.
- 4.4: Prüfkriterien für die Reinigungswirksamkeit: Die Reinigungswirksamkeit muss durch Sichtprüfung und quantitativen Proteinnachweis bestimmt werden. Für nichtinvasive Produkte ist nur eine Sichtprüfung erforderlich.
- 4.4.2: Sichtprüfung: Die Sichtprüfung muss das Nichtvorhandensein von sichtbarer Anschmutzung auf allen einsehbaren Oberflächen der Beladung(en) nach der (den) Reinigungsstufe(n) nachweisen.
ANMERKUNG: Angemessene Anforderungen an die Sichtprüfung können Folgendes umfassen:
- definierte Anweisungen für die Untersuchung;
- angemessene Beleuchtung;
- gegebenenfalls Untersuchungshilfen (z. B. beleuchtetes Boroskop mit Vergrösserung);
- Betrachtungsabstand.
Für weitere Informationen zur Sichtprüfung siehe SN EN 13018.
Die Gute Praxis 2022 empfiehlt beispielsweise eine Beleuchtungsstärke von 1000 Lux für die Prüfung der Sauberkeit und Funktionsfähigkeit von Medizinprodukten.
Gemäss der Norm SN EN 13018 muss das mit der Durchführung der Kontrollen beauftragte Personal eine ausreichende Sehkraft nach EN ISO 9712 besitzen. Für die allgemeine Sichtprüfung muss die Weitsichtigkeit mithilfe eines nach EN ISO 8596 normierten Optotyps überprüft werden. Dabei muss die mit mindestens einem Auge mit oder ohne Korrektur gemessene Sehschärfe mindestens 0,63 betragen. Die Sehkraft muss mindestens alle zwölf Monate überprüft werden.
Diese Prüfung findet heute in den ZSVA nicht statt, aber eine Untersuchung dürfte zeigen, dass sich dank dieser Massnahme das Risiko einer Verwendung nicht konformen Materials (schmutzige Instrumente, kleine Löcher in der Verpackung etc.) verringern lässt.
- 4.4.3: Analysenkriterien. Die Akzeptanzkriterien für die Analyten werden in Form einer Warngrenze sowie einer Eingriffsgrenze festgeschrieben.
- Protein
- Warngrenze ≥ 3 µg/cm2
- Eingriffsgrenze ≥ 6,4 µg/cm2
- Die Analyseverfahren werden in Anhang
- Gesamter organischer Kohlenstoff (TOC)
- Warngrenze ≥ 6 µg/cm2
- Eingriffsgrenze ≥ 12 µg/cm2
- Kohlenhydrat
- Warngrenze ≥ 0,9 µg/cm2
- Eingriffsgrenze ≥ 1,8 µg/cm2
- Ausserdem enthält die Norm Werte für Hämoglobin, ATP und Endotoxin.
- Protein
- Anhang B: Beurteilung der Leistung von proteinbasierten Prüfanschmutzungen: In diesem Anhang werden Prüfverfahren und Prüfeinrichtungen für die Prüfung von Anschmutzungen beschrieben.
Die Schweizerische Leitlinie für die Validierung von Reinigungs- und Desinfektionsprozessen für Medizinprodukte enthält bereits Werte für Proteine, aber nicht für andere Analyten.
Da die Prüfungen nun beschrieben sind, muss von den Lieferanten von Anschmutzungen eine Bescheinigung über die Einhaltung dieser Norm verlangt werden.
- SN EN ISO 15223-1: Medizinprodukte – Symbole zur Verwendung im Rahmen der vom Hersteller bereitzustellenden Informationen – Teil 1: Allgemeine Anforderungen (10.2021)
Die neuen, in dieser Norm veröffentlichten Symbole, die unsere Tätigkeit betreffen, sind:
Importeur
Vertriebspartner 
Sterilisiert mit VH2O2 ![]()
Einfaches Sterilbarrieresystem ![]()
Doppeltes Sterilbarrieresystem ![]()
Einfaches Sterilbarrieresystem mit innenliegender Schutzverpackung 
Einfaches Sterilbarrieresystem mit äusserer Schutzverpackung 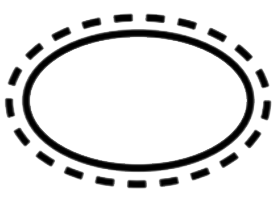
Zur Wiederverwendung an einem einzelnen Patienten 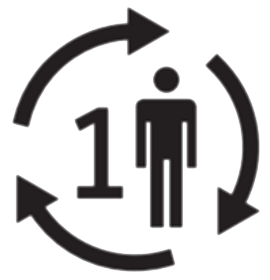
Eindeutige Produktidentifizierung 
SN ISO/TS 16775: Verpackungen für in der Endverpackung zu sterilisierende Medizinprodukte – Leitfaden für die Anwendung von ISO 11607-1 und ISO 11607-2 (11.2021)
-
- Diese Fassung existiert im Gegensatz zu den früheren Versionen auch auf . Der Anhang B, der einen Leitfaden für die Anwendung der Normenreihe ISO 11607 in Einrichtungen des Gesundheitswesens enthält, ist sehr praktisch.
- Darin finden sich die Grundsätze für das Design eines Verpackungssystems, Überlegungen zu Design und Auswahl der Verpackung, Überlegungen zur Zusammenstellung und ein Kapitel über übliche Arten von Sterilbarrieresystemen.
- Des Weiteren enthält Anhang B ein Kapitel über Sterilisierverpackungen mit den verschiedenen Einschlagtechniken (doppelte Diagonalverpackung, Parallelverpackung/quadratischer Falteinschlag).
- Daneben finden sich Abbildungen für das aseptische Öffnen durch die OP-Pflegekraft im sterilen Arbeitsbereich.
- Je ein Kapitel befasst sich mit der Stabilitätsbewertung von Sterilbarrieresystemen (Lagerdauer) und mit dem Ermöglichen der aseptischen Bereitstellung.
- Für die folgenden Verfahren werden die Validierungsanforderungen (IQ, OQ, QP und Routinekontrollen) für Formungs-, Siegelungs- und Zusammenstellungsprozesse beschrieben:
- Siegelungsprozess: Formgebung und Siegelung von Beuteln, Schläuchen oder Tüten;
- Einschlagprozess: Falten und Einschlagen von Sterilisationsbögen;
- Prozess für wiederverwendbare Behälter: Schliessen von wiederverwendbaren Behältern.
- Die Anhänge J bis M enthalten Beispiele von Datenblättern für die Validierung der verschiedenen Verpackungsarten.
- Anhang N bietet ein Beispiel eines Datenblatts für die Bewertung von Sterilverpackungen durch Endanwender.
Die Validierung der Verpackungen ist eine Anforderung der Guten Praxis 2022. Bis ein Schweizer Leitfaden für die Validierung von Verpackungssystemen für Sterilisationsverfahren veröffentlicht wird, können die in Anhang B der Norm SN ISO/TS 16775 bereits sehr detailliert beschriebenen Anforderungen herangezogen werden.
Leitlinien
Schweizerische Leitlinie für den Transport von verunreinigten und aufbereiteten, wiederverwendbaren Medizinprodukten für Aufbereitungseinheiten (09.2021)
Schweizerische Gute Praxis zur Aufbereitung von Medizinprodukten (01.2022)

Im Zusammenhang mit diesen beiden Leitlinien empfiehlt Ihnen die SGSV, Ihre Praxis zu bewerten sowie die Abweichungen und Risiken zu analysieren und mithilfe eines Aktionsplans auf ein akzeptables Niveau zu senken.
Die Inspektion durch Swissmedic basiert auf der Guten Praxis 2022 und den damit verbundenen Leitlinien, zu denen auch die Leitlinie für den Transport gehört (siehe Anhang 5 der Guten Praxis).
Die verschiedenen Versionen der im Artikel genannten Normen können bei der SNV unter https://www.snv.ch/de/erworben werden.